概要
解説
本邦では第15回日本小児遺伝医学会(1992年)で、愛知県心身障害者コロニー中央病院の山中勗、長屋昌宏らが「精神遅滞、てんかん、特異な顔貌を呈する4症例」の演題で初めて発表した疾患である。その後 1998年のJ Med Genet誌にMowatらが症候群として報告した。2001年に愛知県コロニー発達障害研究所の若松らが染色体転座を伴う同症候群例から染色体2q22のZFHX1B遺伝子のハプロ不全が原因であることを示した。先に提示された症例は、後にZFHX1Bの変異が確認された。
正確な発生頻度は不明であるが、原因遺伝子確定後にコロニー中央病院通院中の方だけでも約10名にZFHX1B変異を認めたことから、極めて稀という疾患ではない。ゲシュタルトや身体的特徴で十分診断可能な症候群である。
症状と検査所見
下記のさまざまな大奇形、小奇形を合併する。
発達発育
出生時の身長体重は正常範囲。その後低身長を示す例が約半数。始歩は3~6歳。精神遅滞は重度であるが、周りの人とコミュニケーションを楽しめる子が多い。頭部顔面
眼間開離、細くて目立つ顎、深くて目立つ眼、目立つ鼻尖、持ち上がった目立つ耳朶(耳朶の特徴は本症に特異的であるが表現に難しい、彫りの深い耳介、折れ曲がった耳朶、変形した大きめの耳朶、赤血球様耳朶などの表現がある)、後方に回転した耳介、離れた薄い眉毛(眉毛の正中内側寄りが上方に向かって生え、内側が濃く見えるため平安美人の眉毛のように見える)、比較的大きな口特徴的な耳朶の形態
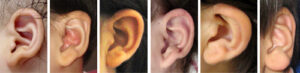
・持ち上がった耳朶 Up-lifted earlobe
・前向きの耳朶 Front facing earlobe
・赤血球様の耳朶(耳朶の中心部の陥凹) Red cell like earlobe
![]()
細い四肢、手指
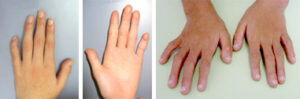
四肢、体格
やせ形の体型、低身長、細長い手指、目立つ指骨間関節、第5指短小
中枢神経系
てんかんを約7割に認める。発作型は多様。小頭症、 脳梁低形成
感覚器
斜視、眼振、反復性中耳炎
循環器
約半数に先天性心疾患を合併。肺動脈狭窄、動脈管開存症、心房中隔欠損症、心室中隔欠損症、ファロー四徴症など多様。
腎泌尿器系
腎泌尿器奇形を約半数に認める。尿道下裂、陰茎彎曲、二分陰嚢、停留精巣、水腎症、膀胱尿管逆流など。
消化器系
30~50%にヒルシュスプルング病が見られ、難治性便秘の合併も多い。
時に見られる症状
口蓋垂裂、粘膜下口蓋裂、気管狭窄、幽門狭窄、小眼球症、虹彩欠損、側弯、歯列不整、膣中隔、嘔吐発作
頻度
正確な頻度は不明である。国内で遺伝子変異確定例が35例、同一知的障害者施設で複数例の報告がある。コロニー中央病院に通院中の子どもの中から10人近くが診断されたことから、実際には診断されていない例が多いと思われる。ヒルシュスプルング病患児の数%とされる。
遺伝子の機能
ZFHX1B(SIP1)は標的遺伝子(CDH1など)のプロモーター領域にあるAGCCT(G)に結合し、転写を抑制する。
遺伝様式・病因
常染色体優性遺伝。染色体2q22のZFHX1B遺伝子のハプロ不全による。ZFHX1B遺伝子単一の変異例が約8割、ZFHX1Bを含む欠失領域を持つ欠失例が約2割。変異型の遺伝子型表現型相関は明かではないが3塩基欠失の軽症例、スプライシング領域変異例や眼球異常の見られるミスセンス変異例など非典型的な表現型が報告されている。欠失例は染色体分染法で診断可能なものからFISH解析可能サイズ以下のものまで様々で、共通の欠失領域は報告されていない。ZFHX1Bを含む欠失では、テロメア側に数Mb以上の欠失が見られる重症例が報告されている(文献5)。生殖腺モザイクによると考えられる同胞例の報告もある(文献6)。男女比はほぼ1:1。
次子再発率
患児のほとんどはde novo(新生変異)であり両親は遺伝子変異を持っていない。生殖腺モザイクによると考えられる同胞例が存在する。次子再発率は生殖腺モザイクの可能性のために一般集団の罹患率より高くなり1~2%と推定される。
経過・治療
診断後は種々の内部奇形の検索が必要である。ヒルシュスプルング病合併例は新生児期から乳児期に根治手術を行う。てんかん合併例は抗てんかん薬で治療。熱性痙攣の合併も多く、痙攣重積例も少なくない。精神運動発達遅滞に対して適切な時期から療育を行い、必要に応じて装具を作成する。運動発達が比較的緩徐なのに対して人への関心やコミュニケーションの意欲が高く、笑いや人との関わりを楽しめる明るい子が多い。遺伝科、小児神経科、眼科、耳鼻科、整形外科、リハビリ科、小児外科など複数科での包括的なフォローアップが必要である。
フォローアップスケジュール
鑑別診断
Goldberg-Shprintzen megacolon 症候群は常染色体劣性遺伝で異なる疾患である。1p36欠失症候群、Angelman症候群、Rubinstein-Taybi症候群、Smith-Lemli-Opitz症候群など。ヒルシュスプルング病が無い場合は難治性便秘が多い。小頭症と顔頭部では眼周囲、尖った下顎と耳介下部の変形が診断の参考になる。耳介の変形(耳介下部のの折れ曲がり、耳朶の前方への膨らみ)は特に本症に特異的であり診断の手がかりとなる。
参考文献
1) Mowat DR, Croaker GDH, Wilson MJ et al :
Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet 35: 617-623, 1998.2) Wakamatsu N, Yamada Y, Nagaya M et al
Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nature Genet. 27: 369-370, 2001.3) Mowat, D. R.; Wilson, M. J.; Goossens, M. :
Mowat-Wilson syndrome. J. Med. Genet. 40: 305-310, 2003.4) Zweier, C.; Temple, I. K.; Beemer, F.; Zackai, E.; Lerman-Sagie, T.; Weschke, B.; Anderson, C. E.; Rauch, A. :
Characterisation of deletions of the ZFHX1B region and genotype-phenotype analysis in Mowat-Wilson syndrome. J. Med. Genet. 40: 601-605, 2003.5) N Ishihara, K Yamada, N Wakamatsu et al
Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22-q24.1:J Med Genet 2004;41:387?3936) Motoko Ohtsuka, Hirokazu Oguni, Mari Matsuo, Makiko Osawa, Kayoko Saito, Yasukazu Yamada, Nobuaki Wakamatsu
J Child Neurology 23(3) 274-278, 2008
Mowat Wilson Sydrome affecting 3 siblings
文責 小児内科 水野誠司